 |
||||||||||
Date: December 5, 2026
by Chaya Venkat

For most of us, the mind-bending and frustrating part of CLL is its diversity. No two patients seem to progress exactly alike, no two seem to respond to therapy in the same way. Until very recently, it was indeed an unfathomable mystery and the once popular "Watch & Wait" approach was the correct answer. When you don't know what you are doing, very often the prudent thing to do is to do nothing. We are still nowhere close to knowing all the answers, but there is enough information trickling through that we can begin to make some good, educated guesses. If after reading "What Type of CLL Do You Have?" and looking over the grim statistics of Bucket C, you scared yourself silly either because you are already there or "Clonal Evolution" may put you in this bucket some time in the future, this article is for you. The science is not easy, but I will try my best to make it easier to understand. This much I can guarantee: the time you spend coming up the learning curve on this and its sister article (to be published in the next day or so) will be well spent, it might make all the difference in how you handle your CLL.
One of the most important items on the check-list we have for putting CLL patients into different risk categories is FISH analysis. Without worrying too much about exactly how this is done, FISH analyzes your specific CLL cells for chromosomal damage. Some forms of damage are more dangerous than others. In this article I would like to focus on two specific chromosomal aberrations that are considered the most dangerous in CLL, the 11q deletion and the 17p deletion. Either of these deletions in the chromosomes of your CLL cells is sufficient to make you a good candidate for the dreaded Bucket C. I am gradually coming around to the point of view that chromosomal aberrations is the name of the game, the specific defect you have in your chromosomes trumps most of the other prognostic indicators. In fact, most of the other prognostic indicators derive their status because of they reflect the fundamental nature of your brand of cancer.
"11q22.3" deletion (11q for short) refers to deletion of a snippet of DNA from chromosome 11 of your DNA. What makes 11q an important deletion in CLL (actually, it is important in most cancers, more on that later on) is that it is the location of a gene called the ATM gene. This gene has very important functions to perform, and when it goes AWOL, all hell breaks loose. The other deletion that puts you in Bucket C is 17p13.1 deletion (17p for short), deletion of a very crucial piece of DNA from your 17th chromosome. This is the location of the P53 gene, also known as the TP53 gene, an important tumor suppressor gene. Defects in the functioning of the ATM gene and TP53 gene are at the heart of most cancers, and in the case of CLL, deletion of either of these two genes meant big trouble. Please note the tense I used, as in the past tense. I plead guilty to charges of being an optimist, but I truly believe that the horrendous statistics sited by Dohner et. al. for 11q or 17p deletions in CLL are no longer quite valid. The research represented in that paper is among the best I have seen, and it has made it possible for us to start thinking of CLL in terms of the fundamental cytogenetic abnormalities that control the disease, but the results are out of date now. For starters, that paper was published in 1997, based on survival statistics of patients treated prior to that date. In other words, pre- monoclonal antibody days, prior to the advent of Rituxan and Campath, just to name two drugs that have changed the ball game. But if you are to get the full benefit of these newer drugs, I think you need to learn a little bit about how ATM and TP53 work, and what goes wrong when these genes are defective in some way. It is hard to come up with solutions without first understanding the problem.
ATM and TP53 are two genes that are woven into the very fabric of your body, into the DNA of each and every cell. These two genes control life and death decisions of the cell, and there is a great deal of interaction between these two genes. Let us consider one cell in your body, that has somehow become damaged. Perhaps it was hit by a stray and random burst of radiation that broke the complex DNA double strand. Perhaps it is just getting very old and senile, falling apart at the seams in the natural course of events. Perhaps it is damaged because some one has fed it a great big dose of chemotherapy drug in an attempt to kill it. For whatever reason, the cell is not quite up to par, and the decision has to be made whether it is time to die or try and repair the damage.
That decision, the thumbs up or thumbs down, is made by ATM and TP53 genes. The question comes up first before the ATM gene. For the sake of an easy analogy, think of the ATM gene as the judge in juvenile court. The damaged cell whose fate hangs in the balance is the defendant in front of the judge, a badly damaged 16 year old who has become a danger to himself and society. There are two choices before Judge ATM: try and rehabilitate the messed up teenager, fix him up to become a model citizen, go forth and raise a family and live happily; or decide that he should be tried as an adult for his crimes, perhaps found guilty and sent off to be executed. Nature hates to waste anything, if it can be saved and used in some fashion. The Judge ATM makes every effort to repair the damaged teen, but just in case the situation is too serious, he also makes sure his back is covered by alerting his superior, Judge TP53 in the adult court. Judge TP53 has the last say in the matter, whether to pronounce the death sentence or let Judge ATM's efforts at rehabilitation have a chance.
Below is a flowchart of how things are supposed to work, in a normal cell that has a well functioning ATM and TP53 genes.
Normal Cell Function
Both ATM and P53 Present
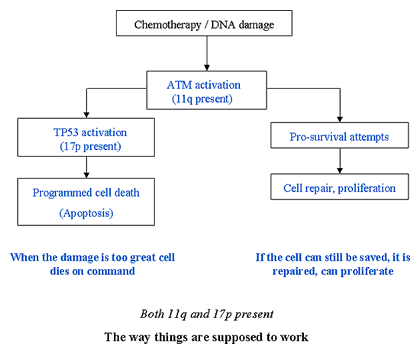
ATM gene is activated as soon as the cell is damaged, and it sets in motion both the cellular repair job as well as the heads-up activation of TP53. If the damage is too great, TP53 gives out the kill signal, and blocks the repair efforts of ATM gene. If the damage can be managed and repaired, ATM's efforts at repair are facilitated and the cell is allowed to live, prosper and proliferate. All is as it should be, no waste of a salvageable cell, and no coddling of a cell damaged beyond the point where it can be safely returned to the bosom of society.
Now let us consider what happens when it is a CLL cell, and you have been trying to kill it by feeding it fludarabine. This chemotherapy drug is a potent poison, it induces DNA damage and breaks the precious double strands of the DNA. No question about it, the damage is real, and very important. If the ATM and TP53 genes are working right, there is no question but the decision would be to let the cell die. Patients in Bucket A, with no defects in ATM or TP53 function, would respond well to fludarabine, since the CLL cell damaged by the chemotherapy dies obediently.
Cell Function with ATM Deletion
Chemoresistance and Tumor Growth
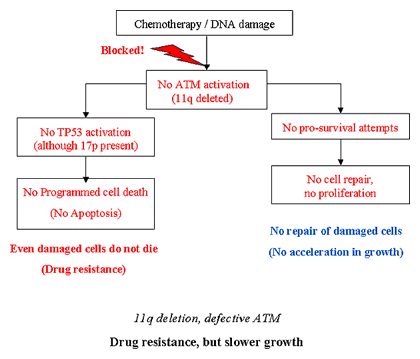
What if ATM function is defective in this cell? The flowchart above explains what may happen. Since Judge ATM is no longer there to do his job, there is no one to attend to the question of what to do with the damage. Judge TP53 is not alerted, no one in charge to make a sensible decision to let this cell die. More often than not, the cell manages to survive, more crippled and malfunctioning than ever. Patients in Bucket C do not respond well to chemotherapy such as fludarabine, and in fact it may lead to CLL cells surviving with even more accumulated mutations as a result of the chemotherapy. In fact, patients with ATM deletion are likely to be candidates for clonal evolution, accumulating more chromosomal abnormalities and secondary cancers as time goes on.
Cell Death Differ. 2026 Apr;10(4):477-84.
Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy.
Sturm I, Bosanquet AG, Hermann S, Guner D, Dorken B, Daniel PT.
Department of Hematology, Oncology and Tumor Immunology, Charite-Campus Berlin-Buch, Humboldt University, 13125 Berlin-Buch, Germany.
Inactivation of p53 has been shown to correlate with poor prognosis and drug resistance in malignant tumors. Nevertheless, few reports have directly shown such effects in primary tumor cells. Here, we investigated the p53 mutational status in 138 B-CLL samples and compared these findings with drug and gamma-irradiation sensitivity profiles. p53 mutations resulted not only in a shorter survival but, notably also in selective resistance to alkylating agents, fludarabine and gamma-irradiation. In contrast, no such effect was observed for vincristine, anthracyclines and glucocorticoids. Thus, these latter compounds induce cell death at least in part by p53-independent pathways. Interestingly, p53 mutations clustered in patients who had received prior chemotherapy. In fact, we show for the first time that treatment with DNA-damaging alkylating agents correlates with occurrence of p53 mutations in a clinical setting. This finding may explain at least to some extent the development of resistance to second-line anticancer chemotherapy.
PMID: 12719725
____________
If you thought that was a bad scene, let us see what happens when the chromosomal defect is not in the ATM gene but the TP53 gene.
Cell Function with P53 Deletion
Chemoresistance and Rapid Tumor Growth
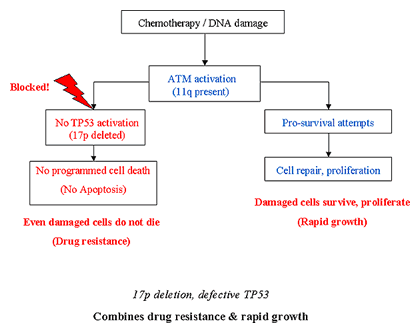
Once again, the CLL cell is fed fludarabine, heavily damaged by the chemotherapy poison, and the question comes up before Judge ATM. This time around our soft-hearted judge has not gone AWOL, he is ready to do his bit. He gets the repair functions geared up, to try and patch up the cell so that it is once again able to resume living and having babies; he also does his duty and informs his superior, TP53. The big problem is that Judge TP53 is not there to ride herd, to make sure that the gentler nature of Judge ATM is not allowed to get away with too much. What can happen in this scenario is that not only is the death sentence not handed down by the malfunctioning TP53, the chemotherapy damaged CLL cell is actually helped to patch itself up and given the direction to go forth and multiply. Patients with TP53 defects do not respond well to chemotherapy, and they also have a rapidly growing disease since damaged cancer cells are efficiently repaired and told to live long and prosper by the well-meaning if dangerous Judge ATM. This is the worst case scenario, no strict TP53 enforce the rules, and efficient ATM doing its bit to repair cell damage in cases where the death penalty is appropriate. Consequently, B-CLL cells in which p53 is non-functional but in which ATM is still active, are not only resistant to chemotherapy but also receive additional ‘survive and proliferate’ signals through ATM activation.
Blood. 1995 Mar 15;85(6):1580-9.
p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias.
Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, Diehl D, Schlenk R, Coy J, Stilgenbauer S, et al.
Medizinische Klinik, University of Heidelberg, Germany.
Conventional cytogenetic analysis in B-cell chronic lymphocytic leukemia (B-CLL) has been very difficult, and the prognostic significance of specific chromosome aberrations is under discussion. Recent improvements in fluorescence in situ hybridization (ISH) techniques have provided an alternative approach for the detection of chromosome aberrations. Here, an interphase cytogenetic study was performed to analyze the incidence and prognostic significance of a p53 gene deletion in B-CLL and related disorders. We studied mononuclear cells from 100 patients with chronic B-cell leukemias [B-CLL, 90 patients; B-prolymphocytic leukemia (B-PLL), 7; Waldenstrom's macroglobulinemia (WM), 3] by fluorescence ISH with a genomic p53 DNA probe. In a subset of patients, additional G-banding analysis and single strand conformation polymorphism (SSCP) analysis was performed. Seventeen of the 100 patients [17%; B-CLL, 11 of 90 (12%); WM, 1 of 3; B-PLL, 5 of 7] exhibited a monoallelic p53 gene deletion by ISH. G-banding analysis demonstrated abnormalities of chromosome 17 in 13 of these 17 patients, all leading to loss of band 17p13. SSCP analysis showed aberrant bands in 9 of 14 patients with a p53 gene deletion. None of 12 patients with a p53 gene deletion compared with 20 of 36 patients (56%) without a deletion responded to therapy with fludarabine or pentostatin (P < .001). The difference in survival probabilities from the time of diagnosis and from the start of treatment with purine analogs between the two groups was highly significant (P < .001). In multivariate analysis, p53 gene deletion was the strongest prognostic factor for survival. In conclusion, p53 gene deletion predicts for non-response to therapy with purine analogs and for poor survival in chronic B-cell leukemias.
PMID: 7888675
___________
The time to treatment and overall survival statistics reported by Dohner et. al. (Genetic Abnormalities in Blood Cancers) are summarized in the table below, and reflect what we have learned thus far. But do remember, these statistics are based on available chemotherapy drugs prior to 1997, before the advent of the monoclonals. You will see how that changes everything, in my opinion, for these higher risk patients.
| Karyotype | Time to Treatment | Overall Survival |
| 17p13 | 9 | 32 |
| 11q23 | 13 | 79 |
| Trisomy 12 | 33 | 114 |
| Normal | 49 | 111 |
| 13q14 | 92 | 133 |
Analogies are great at getting across simple ideas, but they can only go so far. ATM and TP53 function is a complex and very elaborate mechanism developed by our bodies to make sure severely damaged cells are not allowed to survive and create problems for the rest of the body, and at the same time, prevent wasteful death of cells that can be salvaged and repaired. ATM is the major pathway to the activation of TP53 gene, and defects in ATM function have a major effect on function of TP53 as well. But there are suggestions that other pathways may emerge for the activation of TP53, even in the absence of ATM function. This is part of the failsafe and redundancy that the body tries to build into everything. So, in patients with ATM deletion, TP53 may still work to some degree, may be just not as efficiently as it does with its partner ATM pulling its weight. Below is the latest article on the subject, from a credible source. If you are interested in reading the full paper (50 pages of fascinating research), write to us using the Feedback option.
Blood. 2026 Sep 4
Microarray analysis reveals that TP53 and ATM mutant B-CLLs share a defect in activation of pro-apoptotic responses following DNA damage but are distinguished by major differences in activation of pro-survival responses.
Stankovic T, Hubank M, Cronin D, Stewart GS, Fletcher D, Bignell CR, Alvi AJ, Austen B, Weston VJ, Fegan C, Byrd PJ, Moss PA, Taylor AM.
Cancer Research UK Institute for Cancer Studies, University of Birmingham, Birmingham, United Kingdom.
The ATM/p53 dependent DNA damage response pathway plays an important role in the progression of lymphoid tumours. Inactivation of the ATM or TP53 gene is frequent in B cell lymphocytic leukaemia (B- CLL) and leads to aggressive disease. Although the ATM and p53 pathways are overlapping they are not congruent and it is not clear how the mechanism of tumour progression differs between ATM and p53 deficient tumours. Using microarray analysis of ATM mutant, TP53 mutant and ATM/TP53 wild type B-CLLs we show that following exposure to DNA damage transcriptional responses are entirely dependent on ATM function. The p53 pro-apoptotic responses comprise only a part of ATM regulated transcription and additionally ATM regulates pro-survival responses independently of p53. Consequently, the greater severity of the TP53 mutant B-CLLs compared with ATM mutant B-CLLs is consistent with the additive effect of defective apoptotic and elevated survival responses following DNA damage in these tumours. We also show that transcription expression profiles of ATM deficient, TP53 deficient and wild type B-CLLs are indistinguishable before irradiation. Therefore, damage induced transcriptional fingerprinting can be used to stratify tumours according to their biological differences and simultaneously identify potential targets for treatment of refractory tumours.
PMID: 12958068
____________
So far we have talked of deletions as the only reason for genes not doing their job. Sometimes the ATM or TP53 may still be physically present, just not doing their job with any level of efficiency. There is a great deal of work going on in the area of epigenetic control of genes, where the level of efficiency with which a gene works is dependent on what else is happening to it. For example, sometimes it is possible for the TP53 gene to get covered over with a lot of methyl groups (think of it as the gene getting buried under a lot of cellular debris) to the point where it stops functioning. Remove this huge accumulation of methyl groups, and the gene may start working fine again. Hypermethylation is one of the causes of silencing of TP53 gene, and if it can be reversed by stripping away the methyl groups by means of therapy, proper function will be restored. Guess what, some common dietary phytochemicals can do just that!
In my next article I will discuss some things you can do by way of chemoprevention and therapy choices to improve your odds if you happen to be in Bucket C. No kidding folks, this is one area where the more you learn and understand, the better your chances. Perhaps we should include the determination to learn and take charge of your own healthcare as one of the more important prognostic indicators for CLL.
 Enter Keywords: |
———
Disclaimer: The content of this website is intended for information only and is NOT meant to be medical advice. Please be sure to consult and follow the advice of your doctors on all medical matters.
Copyright Notice:
Copyright © 2026-2007 CLL Topics, Inc. All Rights Reserved.
All materials contained on this site are protected by United States copyright law and may not be reproduced, distributed, transmitted, displayed, published or broadcast without the prior written permission of CLL Topics, Inc. You may not alter or remove any trademark, copyright or other notice from copies of the content.
However, you may download and print material from CLLTopics.org exclusively for your personal, noncommercial use.
———




